



Wenn Licht krank macht
«Ein Leben mit EPP ist die Hölle. Welche Hoffnung haben wir?» Dies ist eine von vielen schockierenden Aussagen von Leuten, die mit der seltenen Erbkrankheit Erythropoetische Protoporphyrie (EPP) leben müssen. Sie bilden zu wenig des roten Blutfarbstoffs Hämoglobin. Dies ist aber nicht ihr Hauptproblem. Vielmehr produziert ein nicht verarbeitetes Vorläufermolekül namens PPIX Chaos, indem es mit Licht reagiert und die Haut von innen her verbrennt.
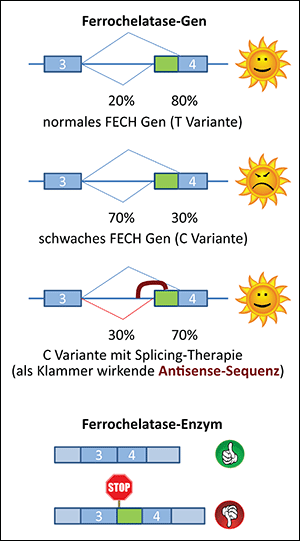
Bei der Erbkrankheit Erythropoetische Protoporphyrie (EPP) wird zu wenig des Enzyms Ferrochelatase gebildet. Patienten besitzen eine Variante des Gens, welche beim Splicen zu wenig korrekte Boten-RNA produziert. Mit der Splicing-Therapie könnte dies korrigiert werden.
Da die Krankeit sehr selten ist, erfahren die meisten Betroffenen erst nach vielen qualvollen Jahren, woran sie leiden. Ein neues Medikament bietet zwar einen teilweisen Schutz gegen die schädlichen Lichtstrahlen, aber eine wirklich wirksame Therapie ist immer noch ausser Sicht.
Der Defekt ist bei den meisten EPP-Patienten (97%) sehr subtil. Sie produzieren etwa 25% des Schlüsselenzyms Ferrochelatase (FECH); 30-40% wären genug, um ein normales Leben zu führen. Der Grund ist, dass diese Personen neben einem defekten FECH Gen eine Variante des normalen Gens haben, die zu wenig Enzym produziert. Durch einen Fehler in der Umsetzung der Information beim sogenannten Splicing erhalten die meisten Boten-RNAs (mRNAs) des Varianten-Gens ein zusätzliches Stück «nonsense»-Information, welches später bei der Proteinsynthese zu einem Kettenabbruch führt.
In einem gemeinsamen Projekt entwickeln unsere Gruppen Therapieansätze, welche dieses fehlerhafte Splicing der FECH mRNA korrigieren sollen.
Unterstützt durch das National Center of Competence in Research (NCCR) RNA & Disease: www.nccr-rna-and-disease.ch.
Beteiligte
Prof. Elisabeth I. Minder
Prof. Xiaoye Schneider-Yin
Dr. des. Jasmin Barman-Aksözen
Prof. Jonathan Hall
Prof. Daniel Schümperli